第Ⅱ相試験:CLEAR-IT Ⅱ試験(用法・用量探索試験;海外データ)1)
1)バイエル薬品社内資料[第Ⅱ相用法・用量探索試験:CLEAR-IT Ⅱ試験(外国人)]承認時評価資料
本剤は海外で実施された第I相試験、第Ⅱ相試験の結果および海外第Ⅲ相試験、日本人を含む第Ⅲ相国際共同試験を基に承認されました。承認時に評価されたデータを紹介しますが、一部国内の承認内容と異なる用法及び用量が含まれています。
CLEAR-IT Ⅱ試験は、滲出型加齢黄斑変性(AMD)患者を対象に、アイリーアの安全性、忍容性、および生物学的作用を検討する目的で実施されました。
試験概要
目的
アイリーアの安全性、忍容性、および生物学的作用を検討する
試験対象
中心窩下CNVを伴う滲出型AMD患者157例(米国33施設)
[主な選択基準]
- 滲出型AMDに伴う中心窩下CNVによる視力低下を有する50歳以上の男女
- OCTによる網膜中心部(病変)の厚さ(CR/LT)が300μm以上
- ETDRS視力表による最高矯正視力文字数が73~34文字
- FAによる病変の最大直線径が5,400μm以下
- 網膜下出血が病変の大きさの50%以下で中心窩にはないこと
- 瘢痕面積が病変の大きさの25%以下 など
試験デザイン
無作為化二重遮蔽試験
投与方法
患者を各群約30例ずつ5群に無作為に割り付け、アイリーア0.5mg、2mgを4週ごとまたは12週ごと、4mgを12週ごとに12週間硝子体内投与した(固定投与期)。続く16~52週目までは、再投与基準に従い、それぞれの用量のPRN(必要に応じ、随時)投与を行った(PRN投与期)。PRN投与の要否は4週ごとに検討した。

[PRN投与期の再投与基準]
- OCTで中心網膜厚(CRT)が100μm以上増加
- OCTで網膜に貯留液が認められ、かつ最高矯正視力文字数が5文字以上低下
- OCTにより検出される遷延性の網膜の貯留液
- 新たなclassic型新生血管
- FAにより検出される新規または遷延性の漏出性変化
- 新たな黄斑部の出血
主な有効性評価項目
主要評価項目
- 12週目におけるOCTによるCR/LTのベースライン値からの変化量
副次評価項目
- ETDRS視力表による最高矯正視力文字数の変化量、視力が改善(最高矯正視力文字数の増加が15文字以上)した患者の割合、CR/LTのベースライン値からの変化量 など
探索的評価項目
- 視力が維持(最高矯正視力文字数の低下が15文字未満)された患者の割合、CNV病変面積の変化量、NEI VFQ-25合計スコアの変化量 など
主な安全性評価項目
有害事象、副作用、重篤な有害事象、投与中止に至った有害事象、死亡 など
解析計画
探索的な解析※
- 主要評価項目(FAS)
- 副次評価項目(FAS)
- 探索的評価項目(FAS) など
- ※ 各評価項目について、記述統計量を算出した。また適切であれば両側検定によるp値を算出し、α=0.049と比較した。
CNV(choroidal neovascularization):脈絡膜新生血管
OCT(optical coherence tomography):光干渉断層計
FA(fluorescein angiography):フルオレセイン蛍光眼底造影
FAS(full analysis set):最大の解析対象集団
NEI VFQ-25(National Eye Institute 25-item Visual Function Questionnaire):米国国立眼病研究所の25項目からなる視覚機能についてのアンケート
中心窩下脈絡膜新生血管を伴う加齢黄斑変性の用法及び用量
アフリベルセプト(遺伝子組換え)として2mg(0.05mL)を1ヵ月ごとに1回、連続3回(導入期)硝子体内投与する。その後の維持期においては、通常、2ヵ月ごとに1回、硝子体内投与する。なお、症状により投与間隔を適宜調節するが、1ヵ月以上あけること。
試験対象例数と各群の患者の内訳

患者背景および特性(FAS)

例数(%)または平均値±標準偏差
- ※1
- 白人/黒人/アジア人以外の人種
- ※2
- classicを含む
試験結果
【形態学的評価】
網膜中心部(病変)の厚さ(CR/LT)
CR/LTの減少は1週目までに投与群全体で認められました。12週目のCR/LTのベースライン値からの変化量は、投与群全体で-118.4±155.2μm(平均値±標準偏差)でした[p<0.0001(名目上のp値):1標本t検定]。また、PRN投与期である52週目では、-129.3±150.4μmでした[p<0.0001(名目上のp値):1標本t検定]。
CR/LTのベースライン値からの変化量(LOCF、FAS):12週(主要評価項目)、12週以外(副次評価項目)
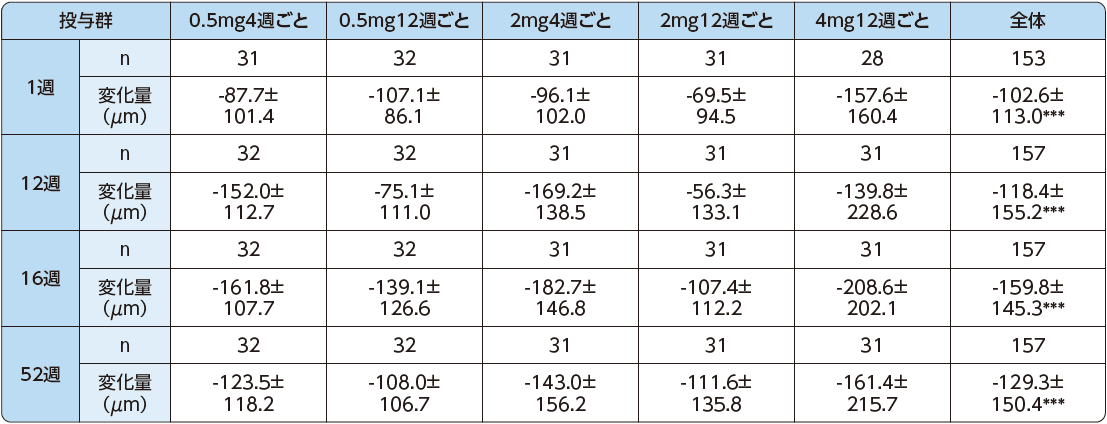
平均値±標準偏差
***:p<0.0001 vs ベースライン値(名目上のp値)(1標本t検定)
【視力評価】
最高矯正視力
最高矯正視力の12週目のベースライン値からの変化量は投与群全体で+5.7±9.9文字(平均値±標準偏差)であり[p<0.0001(名目上のp値):1標本t検定]、0.5mgまたは2mgの4週ごと投与で、それぞれ+8.8±9.2文字、+8.3±10.1文字(平均値±標準偏差)でした。また、PRN投与期である52週目では、+5.3±13.5文字(平均値±標準偏差)でした[p<0.0001(名目上のp値):1標本t検定]。
最高矯正視力文字数の変化量の推移(LOCF、FAS):各観察時点(副次評価項目)

平均値±標準偏差
***:p<0.0001 vs ベースライン値(名目上のp値)(1標本t検定)
本試験の主要評価時点(12週目)において、全体で視力および形態学的な改善が認められました。全体の視力の変化量が+5.7文字であったのに対し、0.5mgと2mgの4週ごと投与群ではそれぞれ+8.8文字、+8.3文字でした。また、4mg投与群では、2mg投与群を上回る効果は認められませんでした。さらに、52週目においても全体で視力および形態学的な改善が認められ、全体の視力の変化量が+5.3文字であったのに対し、固定投与期に2mgの4週ごと投与を受けた群の視力の変化量は+9.0文字でした。8週目の視力の変化量は、各投与群において臨床上明らかな違いはありませんでした。これらのことから、導入期に2mgの4週ごと3回連続投与を行い、その後は8週ごとに投与する方法を第Ⅲ相試験で検討することとしました。
【安全性】
本試験[試験全期間(延長観察期間含む)]におけるすべての有害事象は157例中、全身性で129例(82.2%)、試験眼で134例(85.4%)に認められました。そのうち試験薬に関連する有害事象※は全身性で1例(0.6%)、試験眼で19例(12.1%)でした。
発現率が最も高かった全身性の有害事象は尿路感染16例(10.2%)、試験眼の主な有害事象は結膜出血60例(38.2%)、眼圧上昇30例(19.1%)などでした。全身性の試験薬に関連する有害事象は睡眠障害1例(0.6%)、試験眼の試験薬に関連する主な有害事象は結膜出血8例(5.1%)、眼圧上昇7例(4.5%)などでした。
本試験において、試験薬に関連する重篤な有害事象、試験薬に関連する投与中止に至った有害事象および試験薬に関連する死亡は認められませんでした。
※ 投与手技に関連する有害事象は、試験薬に関連する有害事象に含まれる
ETDRS(early treatment diabetic retinopathy study):糖尿病網膜症早期治療試験
LOCF(last observation carried forward):最終評価スコア外挿法
