SDC達成を見据えたアイリーア8mgによるnAMD治療の可能性
SDC(Sustainable Disease Control)
nAMD (neovascular age-related macular degeneration):滲出型加齢黄斑変性
アイリーア8mgの承認された効能又は効果(抜粋):中心窩下脈絡膜新生血管を伴う加齢黄斑変性
Sustainable Disease Control(SDC)
黄斑疾患治療において、疾患活動性を示す血管新生や血管透過性亢進などの病態を持続してコントロールし、長期的に視力低下を防ぐことが重要であり、SDCとはその治療達成を目指した治療目標です。

監修:鹿児島大学 名誉教授 坂本 泰二 先生
頻回な治療回数、家族や友人の負担への懸念、薬剤にかかる費用負担などは、nAMD患者さんの疾患の負担における主な課題として報告されています(海外データ)
nAMD患者により報告された疾患の負担における主な課題

対象
2021年6月~2022年10月に24ヵ国における77の眼科クリニックで抗VEGF薬による治療中もしくは過去に治療を受けていたnAMD患者4,558例
方法
nAMDの臨床管理についての課題を理解し改善するために、患者の疾患の負担における主な課題などについて、匿名でのアンケートを実施した。リッカート尺度による評価において「強く同意する」「やや同意する」は「同意する」として、「強く同意しない」「やや同意しない」は「同意しない」として統合した。
利益相反
調査の設計、配布、データ収集・管理・分析ならびに本論文の投稿はBayerによって資金提供された。
本論文の著者のうち1名はBayerの社員である。著者にBayer、参天製薬またはRegeneronより講演料、コンサルタント料等を受領している者が含まれる。
Loewenstein A, et al.: Ophthalmol Ther. 2025; 14: 211-228. より作図
日本人を含む第Ⅲ相国際共同試験:PULSAR試験(アフリベルセプト2mgに対する非劣性の検証)
バイエル薬品社内資料[日本人を含む第Ⅲ相国際共同試験:PULSAR試験]承認時評価資料、電子添文改訂時評価資料(96週)
Korobelnik JF, et al.: Ophthalmology. 2025: S0161-6420(25)00532-9. doi: 10.1016/j.ophtha.2025.08.022. Epub ahead of print.
試験概要
【実施地域】アジア太平洋地域(日本含む)、オーストラリア、欧州、中東、南米、北米の27ヵ国、251施設
目的
中心窩下CNVを伴うnAMD患者を対象に、アイリーア8mg12週間隔または16週間隔投与による有効性についてアフリベルセプト2mg8週間隔投与に対する非劣性を検証するとともに、安全性についても検討する
試験対象
中心窩下CNVを伴うnAMD患者1,012例※1(うち日本人:98例)
※1
48および60週目解析では試験薬投与を受けていない1例の患者が無作為割り付け例数に含まれていなかった。
[主な選択基準]
- 試験眼において評価されたnAMDに続発する中心窩下CNVの活動性病変(中心窩に影響を及ぼす傍中心窩病変を含む)を有する50歳以上の男女
- 試験眼の総CNV病変面積(classicおよびoccultの両CNV病変を含む)が病変全体の50%を超える
- 試験眼のETDRS視力表による最高矯正視力文字数が78〜24文字(スネレン視力で20/32〜20/320)であり、nAMDが主な原因であると判断される最高矯正視力文字数の減少がある
- OCTで、試験眼の中心窩領域に影響を及ぼすIRFおよび/またはSRFが認められる など
[主な除外基準]
- 試験眼に、nAMD以外の原因によるCNVを有する
- 試験眼に、蛍光眼底造影により評価した総病変面積が12視神経乳頭面積(12視神経乳頭面積は30.5mm2とし、病変には出血、瘢痕、新生血管を含む)を超える
- 試験眼にコントロール不良の緑内障(抗緑内障薬による治療にもかかわらず眼圧が25mmHgを超える場合)を有する
- 試験眼に特発性または自己免疫性ぶどう膜炎の既往歴を有する
- スクリーニング来院前12週以内に、いずれかの眼に、眼内の炎症または感染を有する
- 試験眼に対する血管新生阻害薬による治療歴を有する
- いずれかの眼における、糖尿病網膜症、糖尿病黄斑浮腫またはnAMD以外の網膜血管疾患の既往歴または臨床所見を有する
- コントロール不良の高血圧(収縮期血圧160mmHg超または拡張期血圧95mmHg超)を有する
- スクリーニング来院前24週以内に脳血管発作または心筋梗塞の既往歴を有する など
試験デザイン
無作為化二重遮蔽実薬対照比較試験
投与方法
対象患者をアフリベルセプト2mg8週間隔投与群、アイリーア8mg12週間隔投与群、アイリーア8mg16週間隔投与群の3群に1:1:1の比で無作為に割り付け※2、硝子体内投与した。試験薬の投与は片眼のみに実施した。
- 2mg8週間隔投与群:アフリベルセプト2mgを4週間隔で連続3回投与後、8週間隔で投与
- 8mg12週間隔投与群:アイリーア8mgを4週間隔で連続3回投与後、12週間隔で投与
- 8mg16週間隔投与群:アイリーア8mgを4週間隔で連続3回投与後、16週間隔で投与
8mg12週間隔投与群および16週間隔投与群の試験薬投与の詳細は、下部の「投与スケジュール、用法用量の変更」を参照のこと。
※2
地域(日本、その他の地域)およびベースラインの最高矯正視力文字数(60文字未満、60文字以上)に基づき層別化した。
主な有効性評価項目
主要評価項目:
48週目における最高矯正視力文字数のベースラインからの変化量
主な副次評価項目:
60週目における最高矯正視力文字数のベースラインからの変化量
16週目に中心窩領域にIRFおよびSRFが認められなかった患者の割合
その他の副次評価項目:
48週目に中心窩領域にIRFおよびSRFが認められなかった患者の割合 など
探索的評価項目:
96週目における最高矯正視力文字数のベースラインからの変化量
8mg12週間隔投与群において96週目まで投与間隔が12週間隔以上であった患者の割合
8mg16週間隔投与群において96週目まで投与間隔が16週間隔以上であった患者の割合
その他の副次評価項目(48週目の評価)に設定した評価項目の96週目の評価 など
主な安全性評価項目
有害事象、副作用、重篤な有害事象、投与中止に至った有害事象、死亡、眼内炎症反応、眼圧上昇事象、眼圧上昇の程度、高血圧事象、APTC定義による動脈血栓塞栓事象 など
事前に規定されたその他の評価項目
8mg12週間隔投与群において96週目に次回予定された投与間隔が12週間隔以上であった患者の割合
8mg16週間隔投与群において96週目に次回予定された投与間隔が16週間隔以上であった患者の割合
48週目および96週目までの投与回数 など
解析計画
主要評価項目および主な副次評価項目において、検定全体のfamily-wiseの第1種の過誤確率を0.025(片側検定)に制御した。主要評価項目および主な副次評価項目における検定の多重性の調整には、下記の階層的検定手順を用い、より上位の階層にランク付けされた仮説を棄却した後にのみ、有意水準0.025(片側)で続く下位の仮説の検定を可能とした※3。
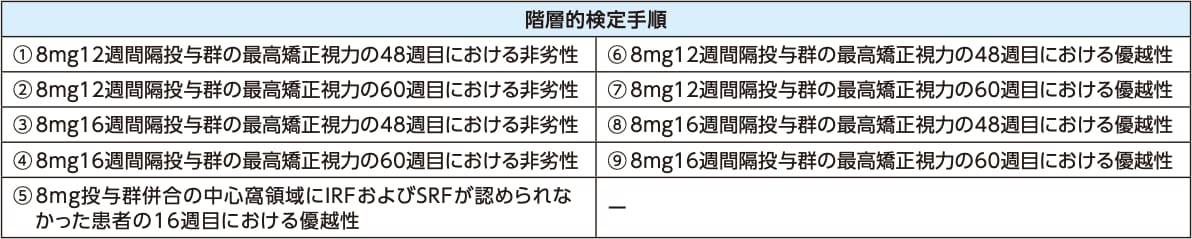
いずれも2mg8週間隔投与群に対する比較検定
階層的検定手順に基づく仮説はすべての患者が60週目を完了(または早期中止)した後に評価した。
主要評価項目を含む48週目までの有効性評価について、60週目完了後のデータによる再解析は実施しなかった。
※3 ⑥において優越性が示されなかったため、検定を終了した。
検証的な解析(第1種の過誤を考慮し、検出力を考慮し例数設計された解析)
主要評価項目(FAS):
8mg12週間隔投与群および8mg16週間隔投与群の2mg8週間隔投与群に対する非劣性の検証(非劣性限界値-4文字)
第1種の過誤を考慮した解析(検出力を考慮した例数設計はされていない)
主な副次評価項目(FAS):
「60週目における最高矯正視力文字数のベースラインからの変化量」は主要評価項目と同一の方法により解析
「16週目に中心窩領域にIRFおよびSRFが認められなかった患者の割合」は8mg投与群併合の2mg8週間隔投与群に対する優越性の検討
探索的な解析
その他の副次評価項目(FAS)、探索的評価項目(FAS、SAF)、事前に規定されたその他の評価項目(SAF)
利益相反
Korobelnik JF, et al.: Ophthalmology. 2025: S0161-6420(25)00532-9. doi: 10.1016/j.ophtha.2025.08.022. Epub ahead of print.
本研究はRegeneronおよびBayerの資金提供により実施され、両社は試験デザイン作成、データ収集、データ解析、原稿作成などに関与した。
著者のうち9名はRegeneronの社員、8名はBayerの社員であり、その他の著者にBayer、Regeneronまたは参天製薬からコンサルタント料、 研究資金等を受領している者が含まれる。
CNV(choroidal neovascularization):脈絡膜新生血管、ETDRS(Early Treatment Diabetic Retinopathy Study):糖尿病網膜症早期治療研究、
OCT(optical coherence tomography):光干渉断層計、IRF(intraretinal fluid):網膜内液、SRF(subretinal fluid):網膜下液、APTC(Antiplatelet Trialists’ Collaboration)
● 中心窩領域:中心窩から直径1mmの範囲
● FAS(full analysis set):最大の解析対象集団。無作為化され、少なくとも1回の試験薬投与を受けたすべての患者。無作為割り付けされた群に基づき解析を行った。
● SAF(safety analysis set):安全性解析対象集団。無作為化され、少なくとも1回の試験薬投与を受けたすべての患者。実際の投与に基づき解析を行った。
投与スケジュール、用法用量の変更
8mg12週間隔投与群および16週間隔投与群では、16週目以降、DRM基準に従い投与間隔を変更した。

DRM基準(短縮:16週目以降):
「最高矯正視力文字数の12週目からの5文字超低下」かつ「CRTの12週目からの25μm超増加、または中心窩に新たな出血、または新たな新生血管が発現」
DRM基準(延長:52週目以降):
「最高矯正視力文字数の12週目からの低下が5文字未満」かつ「OCTで中心窩領域に滲出液が認められない」かつ「中心窩に新たな出血および新生血管の発現がない」
投与間隔の短縮および延長の基準のいずれも満たさなかった患者は投与間隔を維持した。
DRM(dose regimen modification):用法用量変更、CRT(central retinal thickness):中心網膜厚
● 中心網膜厚:中心窩領域の網膜厚
中心窩下脈絡膜新生血管を伴う加齢黄斑変性の用法及び用量
アフリベルセプト(遺伝子組換え)として8mg(0.07mL)を4週ごとに1回、通常、連続3回(導入期)硝子体内投与するが、症状により投与回数を適宜減じる。その後の維持期においては、通常、16週ごとに1回、硝子体内投与する。なお、症状により投与間隔を適宜調節するが、8週以上あけること。
Focus on Vision Gain
主要評価項目
(検証的解析結果)
48週目における最高矯正視力文字数のベースラインからの変化量は、8mg12週間隔投与群で+6.1文字、8mg16週間隔投与群で+5.9文字であり、2mg8週間隔投与群(+7.0文字)に対する非劣性が検証されました
最高矯正視力文字数のベースラインからの変化量(MMRM、FAS)
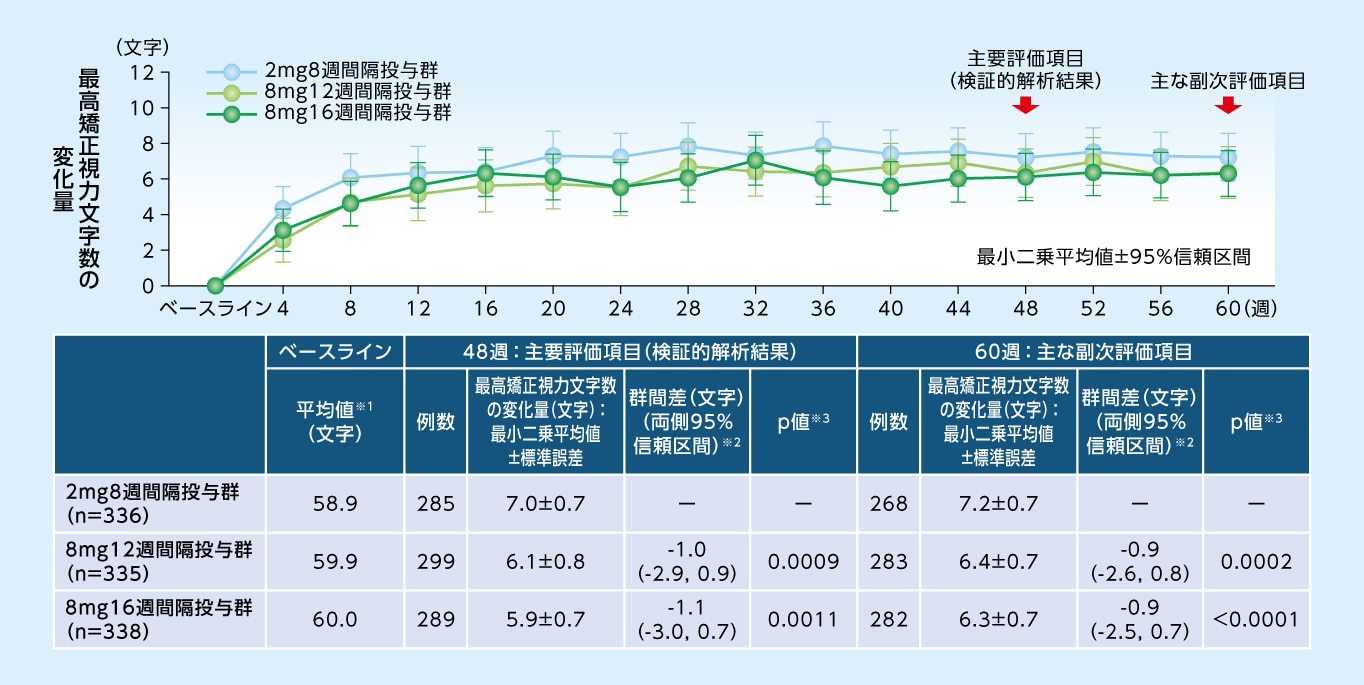
※1 実測値 ※2 各群−2mg8週間隔投与群 ※3 非劣性(非劣性限界値-4文字)の片側検定
階層的検定手順に従い、下位の「8mg投与群併合の中心窩領域にIRFおよびSRFが認められなかった患者の16週目における優越性」は示されたが、続く「8mg12週間隔投与群の最高矯正視力の48週目における優越性」が示されなかったため、検定を終了した。
●
MMRM(mixed model for repeated measurements):反復測定混合効果モデル。ベースラインの最高矯正視力文字数を共変量、投与群、来院および層別因子[地域(日本、その他の地域)、ベースラインの最高矯正視力文字数(60文字未満、60文字以上)]を固定効果とし、ベースラインの最高矯正視力文字数と来院の交互作用項、投与群と来院の交互作用項を含む。
探索的評価項目
96週目における最高矯正視力文字数のベースラインからの変化量は、8mg12週間隔投与群で+5.6文字、8mg16週間隔投与群で+5.5文字、2mg8週間隔投与群で+6.6文字でした
最高矯正視力文字数のベースラインからの変化量(MMRM、FAS)
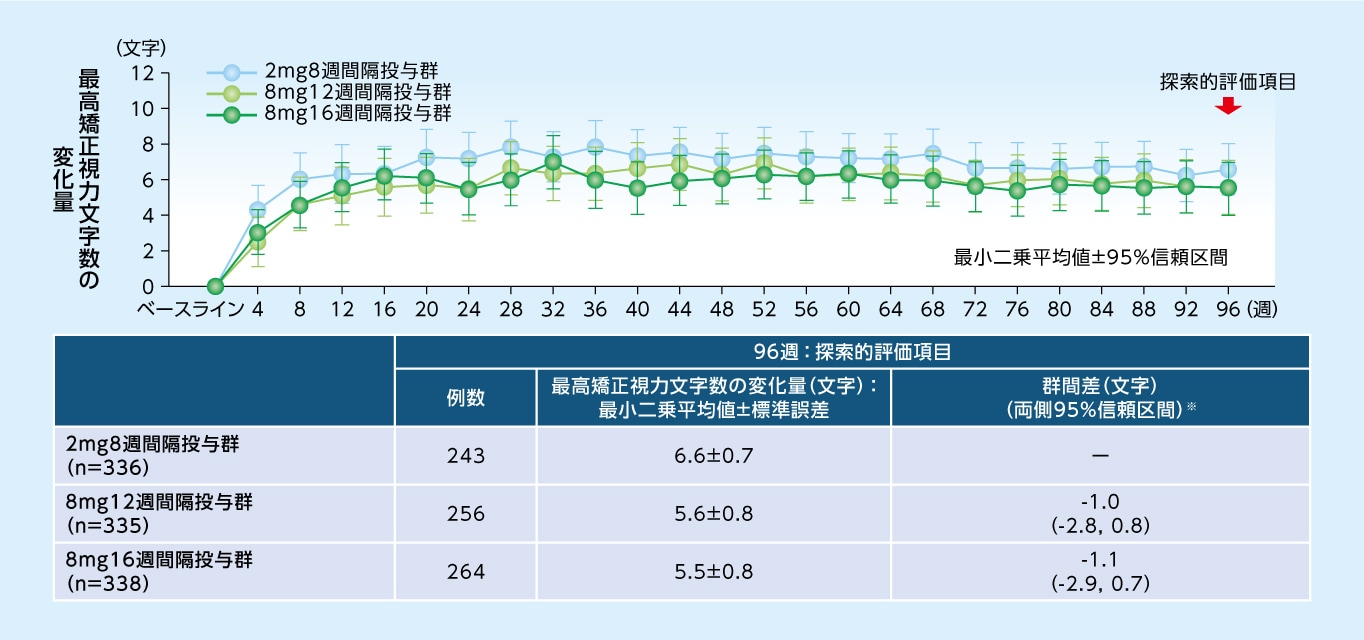
※ 各群ー2mg8週間隔投与群
●
MMRM:ベースラインの最高矯正視力文字数を共変量、投与群、来院および層別因子[地域(日本、その他の地域)、ベースラインの最高矯正視力文字数(60文字未満、 60文字以上)]を固定効果とし、ベースラインの最高矯正視力文字数と来院の交互作用項、投与群と来院の交互作用項を含む。
バイエル薬品社内資料[日本人を含む第Ⅲ相国際共同試験:PULSAR試験]承認時評価資料、電子添文改訂時評価資料(96週)
Focus on Fluid Status
主な
副次評価項目
16週目に中心窩領域にIRFおよびSRFが認められなかった患者の割合は、8mg投与群併合で63.3%であり、2mg8週間隔投与群(51.6%)に対する優越性が示されました
16週目に中心窩領域にIRFおよびSRFが認められなかった患者の割合(LOCF、FAS)
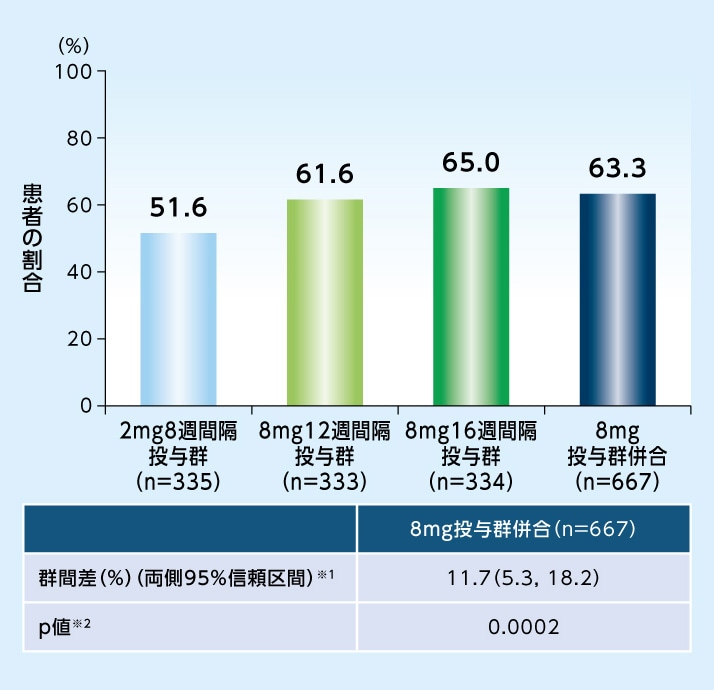
※1 8mg投与群併合−2mg8週間隔投与群[地域(日本、その他の地域)およびベースラインの最高矯正視力文字数(60文字未満、60文字以上)で層別化したMantel- Haenszel型の重みを用いて調整した]
※2 地域(日本、その他の地域)およびベースラインの最高矯正視力文字数(60文字未満、60文字以上)で調整した片側CMH検定
●
LOCF(Last Observation Carried Forward):最終評価スコア外挿法。欠測値に対して欠測前の最後の測定値を用いて補完する解析方法
探索的評価項目
96週目に中心窩領域にIRFおよびSRFが認められなかった患者の割合は、8mg12週間隔投与群で69.6%、8mg16週間隔投与群で63.6%、2mg8週間隔投与群で66.5%でした
中心窩領域にIRFおよびSRFが認められなかった患者の割合(LOCF、FAS)
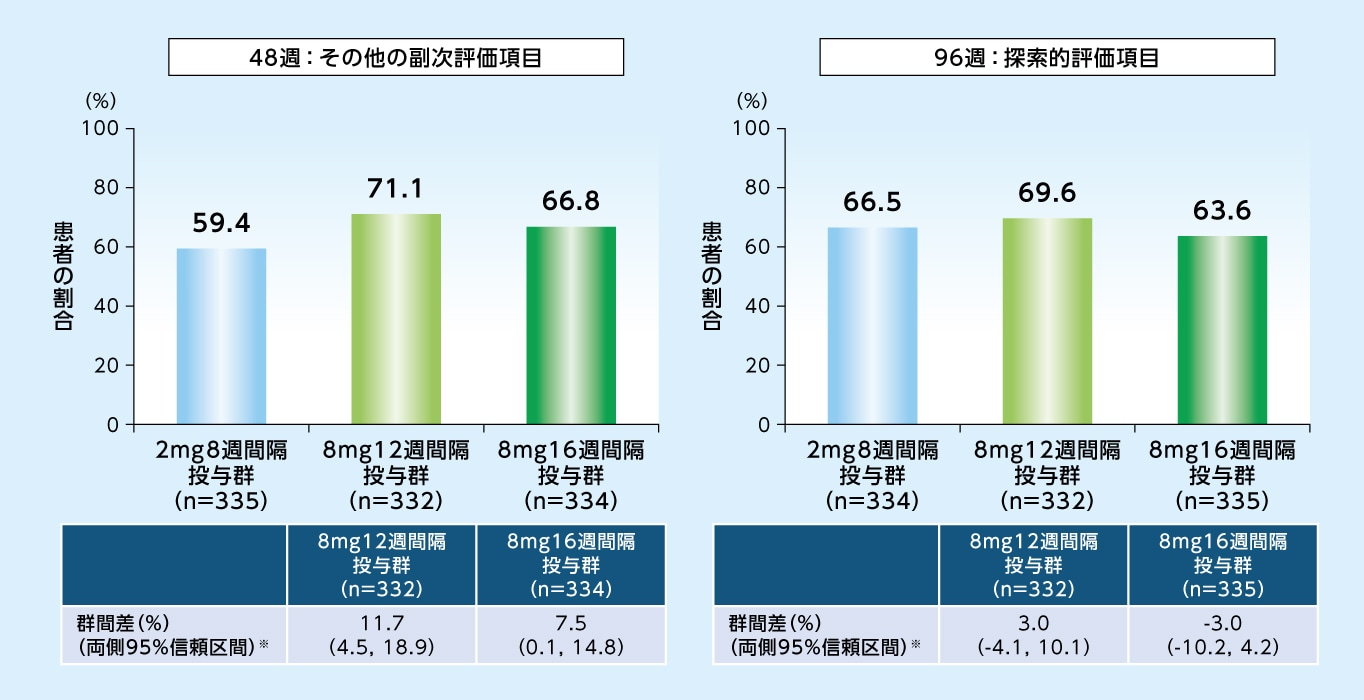
※ 各群-2mg8週間隔投与群[地域(日本、その他の地域)およびベースラインの最高矯正視力文字数(60文字未満、60文字以上)で層別化したMantel-Haenszel型の重みを用いて調整した]
バイエル薬品社内資料[日本人を含む第Ⅲ相国際共同試験:PULSAR試験]承認時評価資料、電子添文改訂時評価資料(96週)
Focus on Treatment Burden
事前に規定された
その他の評価項目
8mg12週間隔投与群において96週目に次回予定された投与間隔が12週間隔以上であった患者の割合は86.6%、20週間隔以上であった患者の割合は40.5%でした
8mg12週間隔投与群において96週目に次回予定された投与間隔別の患者の割合※1(SAF※2)1,2)
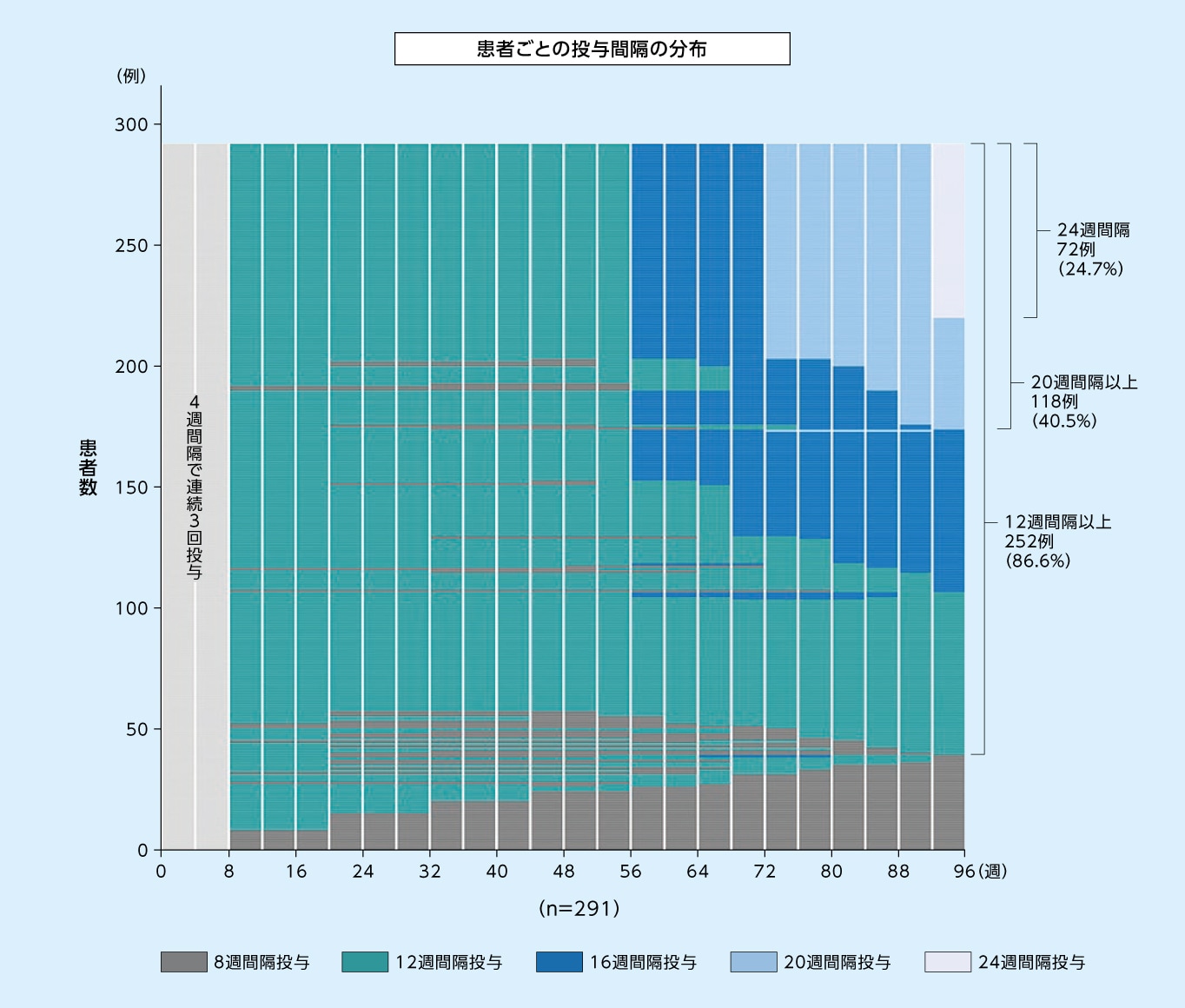
縦の白線は来院を示す
※1 96週目より前の最終来院日における評価に基づく投与間隔(すなわち92週目までの最終来院日における評価に基づく投与間隔)
※2 SAFのうち96週目までの投与を完了した患者のみ
8mg12週間隔投与群において96週目まで投与間隔が12週間隔以上であった患者の割合(SAF※)(探索的評価項目)2)

※ SAFのうち96週目までの投与を完了した患者のみ
1)Korobelnik JF, et al.: Ophthalmology. 2025: S0161-6420(25)00532-9. doi: 10.1016/j.ophtha.2025.08.022. Epub ahead of print. より改変
2)バイエル薬品社内資料[日本人を含む第Ⅲ相国際共同試験:PULSAR試験]電子添文改訂時評価資料(96週)
事前に規定された
その他の評価項目
8mg16週間隔投与群において96週目に次回予定された投与間隔が16週間隔以上であった患者の割合は78.4%、20週間隔以上であった患者の割合は53.1%でした
8mg16週間隔投与群において96週目に次回予定された投与間隔別の患者の割合※1(SAF※2)1,2)
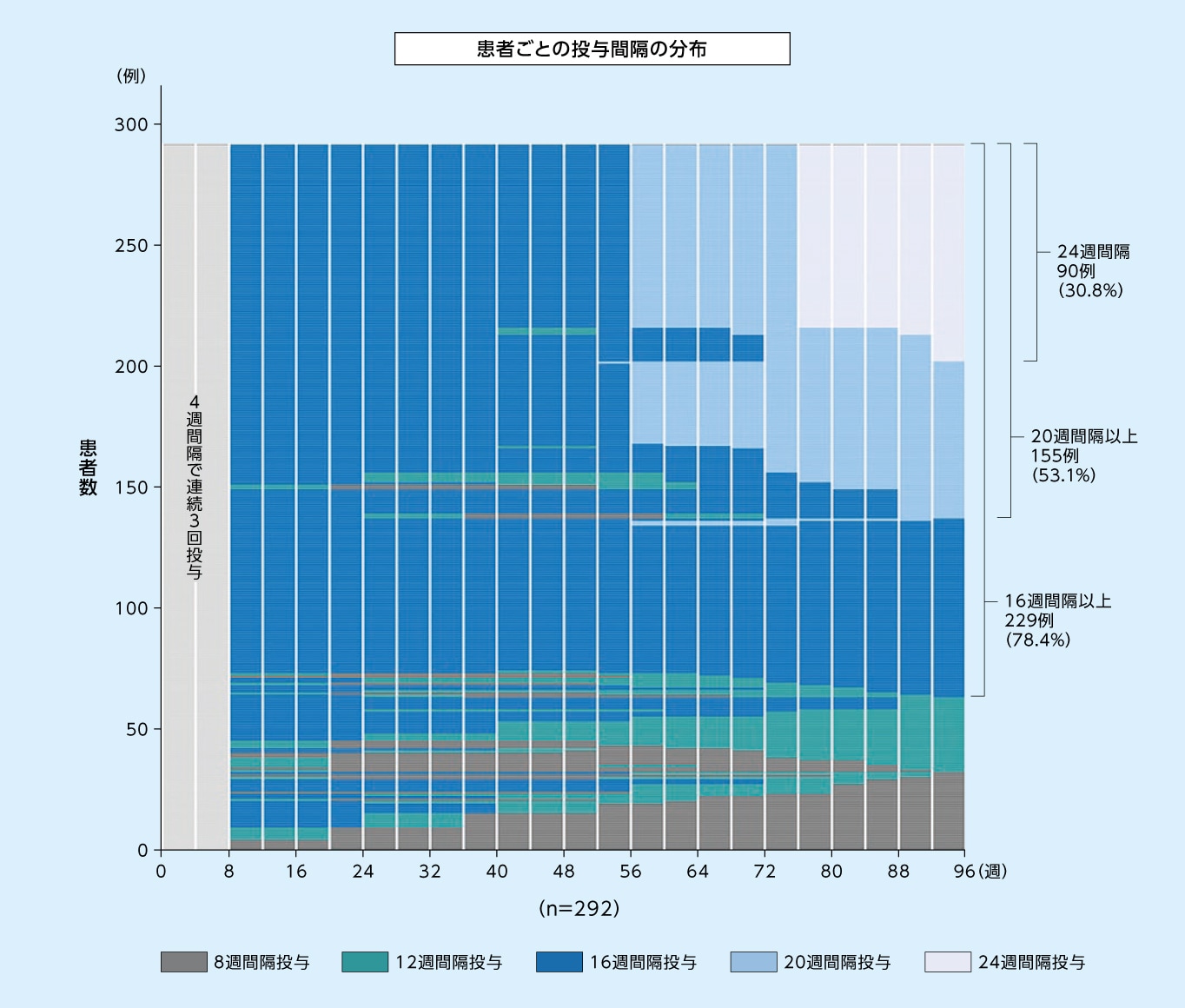
縦の白線は来院を示す
※1 96週目より前の最終来院日における評価に基づく投与間隔(すなわち92週目までの最終来院日における評価に基づく投与間隔)
※2 SAFのうち96週目までの投与を完了した患者のみ
8mg16週間隔投与群において96週目まで投与間隔が16週間隔以上であった患者の割合(SAF※)(探索的評価項目)2)

※ SAFのうち96週目までの投与を完了した患者のみ
1)Korobelnik JF, et al.: Ophthalmology. 2025: S0161-6420(25)00532-9. doi: 10.1016/j.ophtha.2025.08.022. Epub ahead of print. より改変
2)バイエル薬品社内資料[日本人を含む第Ⅲ相国際共同試験:PULSAR試験]電子添文改訂時評価資料(96週)
事前に規定された
その他の評価項目
試験眼に対する96週目までの投与回数は、8mg12週間隔投与群で9.2回、8mg16週間隔投与群で7.8回、2mg8週間隔投与群で11.9回でした
48週目および96週目までの投与回数※1(試験眼、SAF)
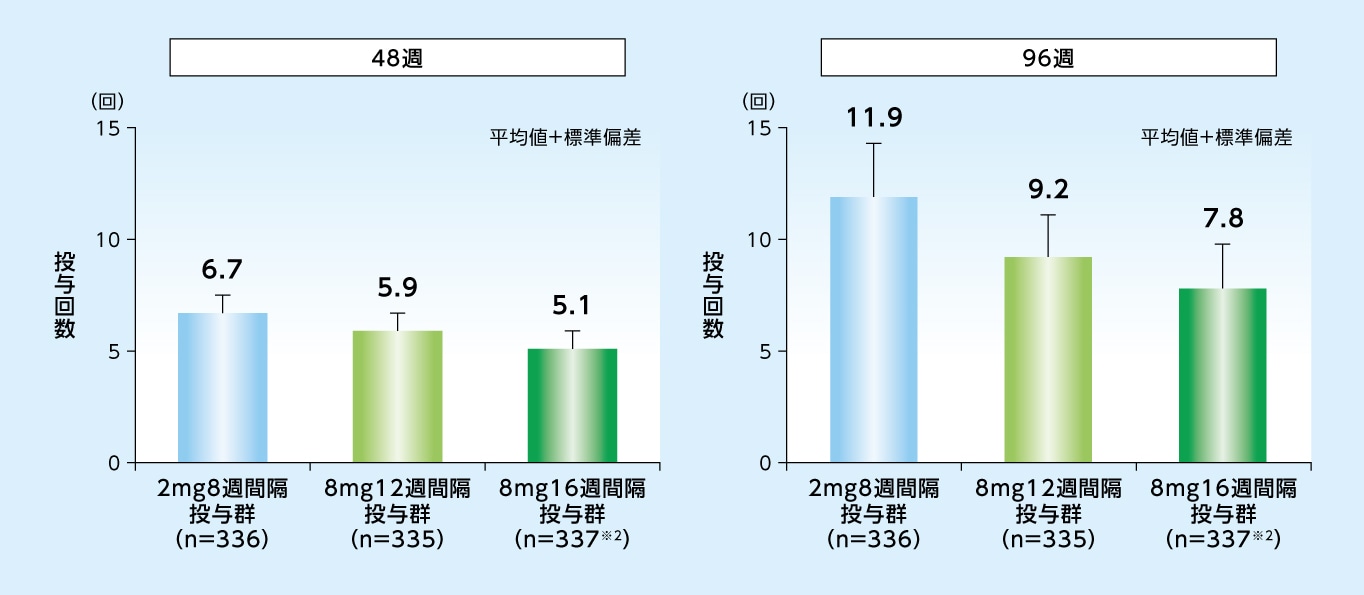
※1 偽注射を除く投与回数
※2 SAF(n=338)のうち1例が欠測
バイエル薬品社内資料[日本人を含む第Ⅲ相国際共同試験:PULSAR試験]承認時評価資料、電子添文改訂時評価資料(96週)
Safety
96週間において、すべての有害事象は8mg12週間隔投与群で335例中292例(87.2%)、8mg16週間隔投与群で338例中303例(89.6%)、2mg8週間隔投与群で336例中300例(89.3%)に認められました
有害事象(96週間、SAF)
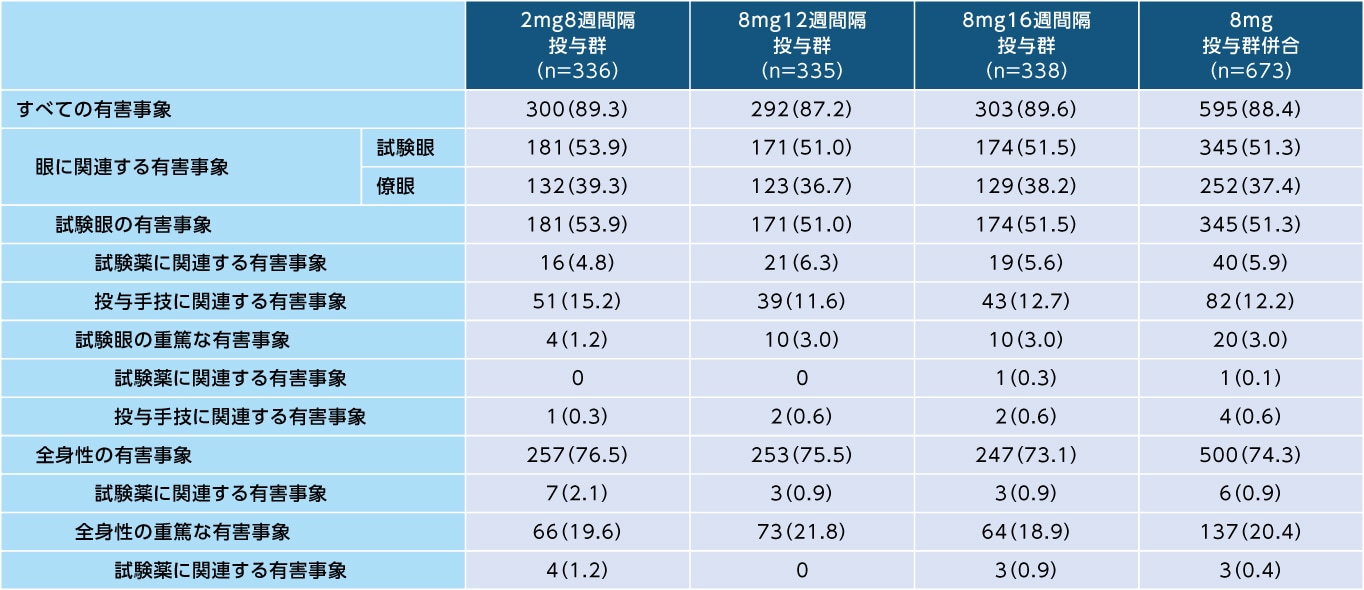
発現例数(発現割合%)
96週間において、各群の主な有害事象、試験薬に関連する重篤な有害事象、試験薬に関連する投与中止に至った有害事象、試験薬に関連する死亡は以下の通りでした
主な有害事象(96週間、SAF)
8mg12週間隔投与群:
COVID-19 58例(17.3%)、白内障31例(9.3%)、高血圧23例(6.9%)、視力低下21例(6.3%)、網膜出血、上咽頭炎、背部痛が各18例(5.4%)、網膜下液14例(4.2%)、尿路感染13例(3.9%)、眼圧上昇、下痢、関節痛が各12例(3.6%)、上気道感染11例(3.3%)、黄斑肥厚、結膜出血、新生血管加齢黄斑変性が各10例(3.0%) など
8mg16週間隔投与群:
COVID-19 72例(21.3%)、白内障32例(9.5%)、上咽頭炎27例(8.0%)、視力低下、高血圧が各23例(6.8%)、網膜出血19例(5.6%)、硝子体浮遊物17例(5.0%)、尿路感染16例(4.7%)、背部痛15例(4.4%)、咳嗽13例(3.8%)、硝子体剥離12例(3.6%)、黄斑浮腫、眼圧上昇、関節痛が各11例(3.3%)、肺炎、変形性関節症が各10例(3.0%) など
2mg8週間隔投与群:
COVID-19 60例(17.9%)、上咽頭炎30例(8.9%)、視力低下24例(7.1%)、白内障、背部痛が各22例(6.5%)、尿路感染21例(6.3%)、網膜出血19例(5.7%)、高血圧18例(5.4%)、網膜下液、硝子体浮遊物が各16例(4.8%)、転倒13例(3.9%)、上気道感染12例(3.6%)、ドライアイ11例(3.3%)、黄斑浮腫、眼圧上昇、気管支炎、咳嗽、発熱が各10例(3.0%) など
試験薬に関連する重篤な有害事象(96週間、SAF)
8mg12週間隔投与群: 本試験においては認められなかった
8mg16週間隔投与群: 心筋梗塞2例、閉塞隅角緑内障、肺塞栓症が各1例
2mg8週間隔投与群: 脳血管発作2例、急性心筋梗塞、高血圧が各1例
試験薬に関連する投与中止に至った有害事象(96週間、SAF)
8mg12週間隔投与群: 本試験においては認められなかった
8mg16週間隔投与群: 虹彩毛様体炎、網膜下液が各1例
2mg8週間隔投与群: 脳血管発作、ぶどう膜炎、血管炎が各1例
試験薬に関連する死亡(96週間、SAF)
本試験においては認められなかった
MedDRA ver.26.0
バイエル薬品社内資料[日本人を含む第Ⅲ相国際共同試験:PULSAR試験]、電子添文改訂時評価資料(96週)
アイリーア高濃度製剤の特徴・臨床試験計画
アイリーア高濃度製剤の特徴
アイリーア8mg(アフリベルセプト濃度114.3mg/mL)
アフリベルセプト濃度を既承認の40mg/mL(用量2mg)※から114.3mg/mL(用量8mg)へ増加することにより、より多くの有効成分の硝子体内投与を可能とした新たな製剤である
アイリーア8mgの硝子体内投与では、アフリベルセプト2mgと比べ作用持続時間の延長が期待できる
アイリーア8mgの臨床試験計画
nAMDおよびDME患者を対象に、アイリーア8mgの12週間隔または16週間隔投与が、アフリベルセプト2mgの8週間隔投与と比較して、同様の有効性および安全性を示すかを検討することを目的とし、臨床試験を計画した
※ 未熟児網膜症以外の効能及び効果における用量[未熟児網膜症:40mg/mL(用量0.4mg)]
バイエル薬品社内資料[海外第Ⅱ相試験:CANDELA試験]承認時参考資料
バイエル薬品社内資料[日本人を含む第Ⅲ相国際共同試験:PULSAR試験]承認時評価資料
バイエル薬品社内資料[日本人を含む第Ⅱ/Ⅲ相国際共同試験:PHOTON試験]承認時評価資料
[PPKモデルに基づくシミュレーション]
アフリベルセプト2mg投与8週後の眼内の遊離型アフリベルセプト濃度(中央値)は、アイリーア8mg投与14週後と同程度と推定されました
アフリベルセプト2mgおよびアイリーア8mg単回硝子体内投与後の眼内の遊離型アフリベルセプト曝露量の
PPKモデルに基づく予測推移(日本人および外国人データ)
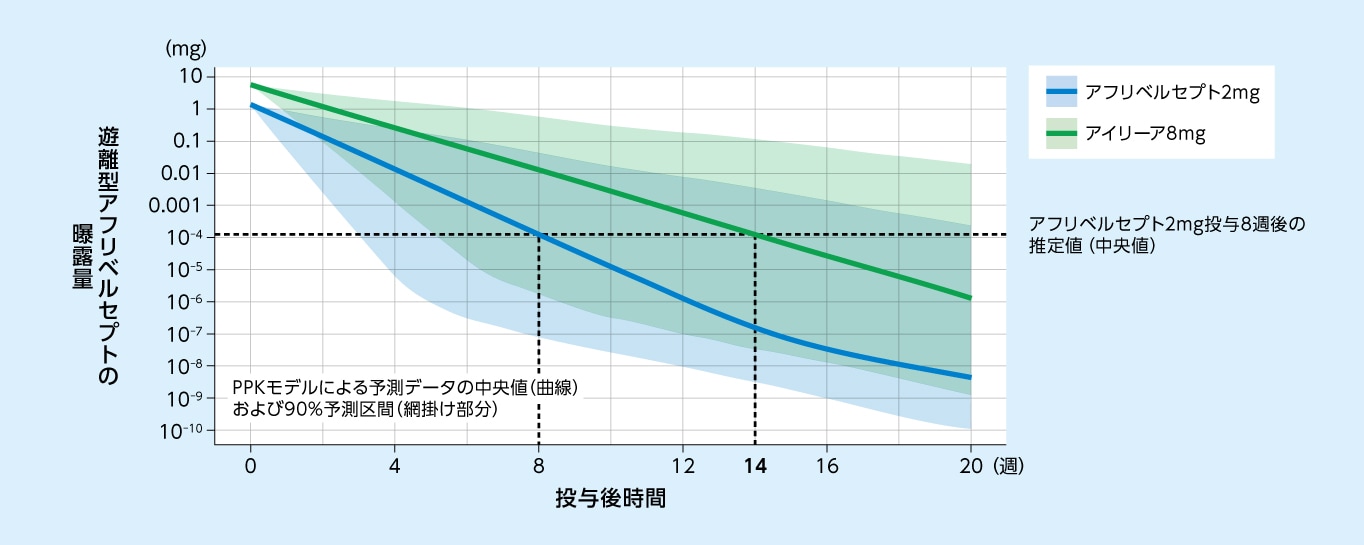
PPKモデル:母集団薬物動態モデル コンパートメントモデル:体内を1つまたは複数の区画(コンパートメント)に分けて単純化したモデル
方法
nAMDおよびDME患者を対象にアイリーア8mgを投与した3試験(CANDELA試験、PULSAR試験、PHOTON試験)に加え、nAMD患者、DME患者、健康成人およびがん患者を対象にアフリベルセプトを投与した13試験の計16試験のデータを用いて、アフリベルセプトのPPKモデル(アフリベルセプトが中心コンパートメントにおいてMichaelis-Menten機構によりVEGFと結合する3-コンパートメントモデル[中心(血漿)および2つの末梢(組織)のコンパートメントにより構成])を構築した。このPPKモデルに基づき、nAMDおよびDME患者の眼にアフリベルセプト2mgおよびアイリーア8mgを単回硝子体内投与したときの遊離型アフリベルセプト曝露量の推移を予測した。
バイエル薬品社内資料[薬物動態:母集団薬物動態解析]アイリーア8mg承認時評価資料
アイリーア8mg:中心窩下脈絡膜新生血管を伴う加齢黄斑変性、糖尿病黄斑浮腫の用法及び用量
アフリベルセプト(遺伝子組換え)として8mg(0.07mL)を4週ごとに1回、通常、連続3回(導入期)硝子体内投与するが、症状により投与回数を適宜減じる。その後の維持期においては、通常、16週ごとに1回、硝子体内投与する。なお、症状により投与間隔を適宜調節するが、8週以上あけること。
PULSAR試験
継続期間(96~156週目)の試験概要・投与スケジュール
バイエル薬品社内資料[日本人を含む第Ⅲ相国際共同試験:PULSAR試験 継続期間]
試験概要
目的
中心窩下CNVを伴うnAMD患者を対象とした主試験(96週目まで)の後の試験継続(156週目まで)を通して、アイリーア8mgの2年を超える投与の長期的な有効性および安全性、ならびにアフリベルセプト2mgからアイリーア8mgに切り替えた際の有効性および安全性を検討する
試験対象
主試験の後の試験継続に同意したすべての患者※:625例
※
主試験における96週目の試験終了時来院を完了し、続けて継続期間(156週目まで)の参加に同意した患者
[主な選択基準]
- 主試験の期間中に試験眼のnAMDに対する試験薬以外の治療が行われていない
- 84、88、92週目のいずれかで少なくとも1回の最高矯正視力文字数およびCRT値が得られている など
投与方法
継続期間(96~156週目)では、すべての患者の試験眼にアイリーア8mgを硝子体内投与した。
- アフリベルセプト2mg8週間隔/アイリーア8mg切り替え投与群:
96週目に最初のアイリーア8mg投与を実施し、初期投与間隔は12週間隔とした。104週目にE-DRM基準(短縮)を満たした場合、
当該来院日に投与を行い、以降は8週間隔に短縮した。その後の投与間隔はE-DRM基準(短縮および延長)に基づき2週幅で調整した。 - アイリーア8mg12週間隔投与群、アイリーア8mg16週間隔投与群:
主試験終了時と同じ投与間隔を継続した。その後の投与間隔はE-DRM基準(短縮および延長)に基づき2週幅で調整した。
E-DRM(extension phase dose regimen modification):継続期間における用法用量変更
投与スケジュール、用法用量の変更:3年目(96~156週目)
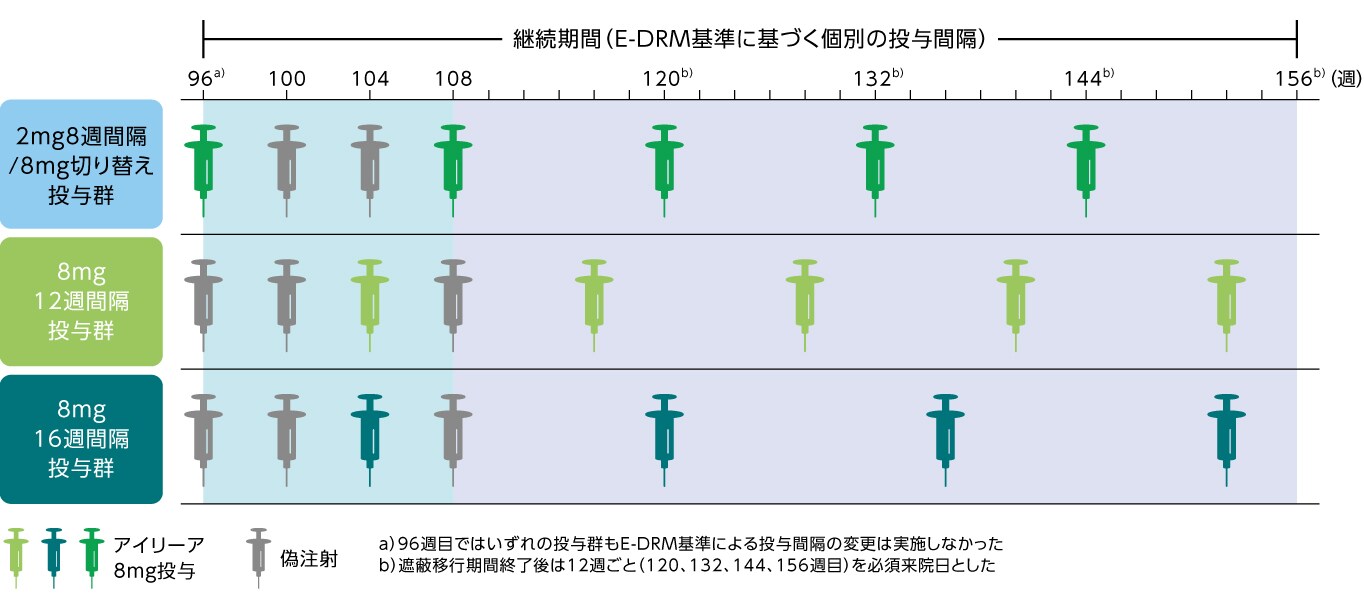
遮蔽移行期間(必須来院4週ごと)
2mg8週間隔/8mg切り替え投与群では96週目に最初のアイリーア8mg投与を実施し、初期投与間隔は12週間隔とした。
ただし、104週目にE-DRM基準(短縮)を満たした場合、当該来院日に投与を行い、以降は8週間隔に短縮した。
8mg12週間隔投与群および16週間隔投与群では主試験終了時と同じ投与間隔を継続した。
非遮蔽パート(必須来院12週ごと)
2mg8週間隔/8mg切り替え投与群では104週目以降、8mg12週間隔投与群および16週間隔投与群では100週目以降、投与 来院であるかを問わず、すべての来院時にE-DRM基準(短縮)を満たすかどうか評価した。
- いずれの来院日においてもE-DRM基準(短縮)を満たした場合、当該来院日に投与を実施した(直近の投与からの経過が8週未満の場合を除く)。
- 投与来院日にE-DRM基準(短縮)を満たした場合、次回の投与間隔を2週幅で短縮した(最短8週間隔)。
- 投与を実施しない来院日にE-DRM基準(短縮)を満たした場合、次回の投与間隔を直近の投与間隔から2週幅で短縮した(最短8週間隔)。
投与間隔の短縮に加えて、すべての患者は投与来院日にE-DRM基準(延長)を満たした場合、次回の投与間隔を2週幅で延長した(最長24週間隔)。
E-DRM基準(短縮):
「nAMDの遷延または悪化による最高矯正視力文字数の新規ベースライン(NB)※からの5文字超低下」かつ
「CRTのNBからの25μm超増加」または「中心窩に新たな出血」または「中心窩に新たな新生血管が発現」
または
「nAMDの悪化による最高矯正視力文字数のNBからの10文字超低下」
E-DRM基準(延長):
「最高矯正視力文字数のNBからの低下が5文字未満」かつ「OCTで中心窩領域に滲出液が認められない」かつ
「中心窩に新たな出血および新生血管の発現がない」
※
継続期間におけるNB:84、88および92週目の最高矯正視力文字数またはCRTの各平均値(端数を切り上げて整数とした)
投与間隔の短縮および延長の基準のいずれも満たさなかった患者は投与間隔を維持した。
まとめ
nAMD患者さんにおいて、疾患の負担における主な課題として、「頻回な治療回数」、「家族や友人の負担への懸念」、「薬剤にかかる費用負担」などを抱えていることが報告されています
黄斑疾患治療において、疾患活動性を示す血管新生や血管透過性亢進などの病態を持続してコントロールし、長期的に視力低下を防ぐことが重要であり、SDCとはその治療達成を目指した治療目標です。

頻回な治療回数、家族や友人の負担への懸念、薬剤にかかる費用負担などは、nAMD患者さんの疾患の負担における主な課題として報告されています(海外データ)1)。
[PULSAR試験]
アイリーア8mgはPULSAR試験において、連続3回の導入期と最短8週最長24週の投与間隔による維持期の設定のもと96週目までの有効性が示されました
導入期からアイリーア8mgでnAMDの治療を開始することでSDCの達成が期待できます
Focus on Vision Gain
96週目における最高矯正視力文字数のベースラインからの変化量(最小二乗平均値)は、8mg12週間隔投与群で+5.6文字、8mg16週間隔投与群で+5.5文字、2mg8週間隔投与群で+6.6文字でした2)。
Focus on Fluid Status
96週目に中心窩領域にIRFおよびSRFが認められなかった患者の割合は、8mg12週間隔投与群で69.6%、8mg16週間隔投与群で63.6%、2mg8週間隔投与群で66.5%でした2)。
Focus on Treatment Burden
8mg16週間隔投与群において96週目まで投与間隔が16週間隔以上であった患者の割合は70.2%であり、96週時点で、53.1%で次回予定された投与間隔を20週間隔以上に、30.8%で24週間隔に延長すると判断されていました2)。
Safety
96週間において、すべての有害事象は8mg12週間隔投与群で335例中292例(87.2%)、8mg16週間隔投与群で338例中303例(89.6%)、2mg8週間隔投与群で336例中300例(89.3%)に認められました2)。
96週間において、主な有害事象は8mg12週間隔投与群でCOVID-19 58例(17.3%)、白内障31例(9.3%)、8mg16週間隔投与群でCOVID-19 72例(21.3%)、白内障32例(9.5%)、2mg8週間隔投与群でCOVID-1960例(17.9%)、上咽頭炎30例(8.9%)などでした2)。
アイリーア8mgは、アフリベルセプト2mgに比べ、投与間隔の延長が期待される薬剤です
PPKモデルに基づくシミュレーションでは、アフリベルセプト2mg投与8週後の眼内の遊離型アフリベルセプト濃度は、アイリーア8mg投与14週後と同程度と推定されました3)。
PULSAR試験 継続期間は以下の検討を目的としています4)
アイリーア8mgの2年を超える投与の長期的な有効性および安全性
アフリベルセプト2mgからアイリーア8mgに切り替えた際の有効性および安全性
1)Loewenstein A, et al.: Ophthalmol Ther. 2025; 14: 211-228.
2)バイエル薬品社内資料[日本人を含む第Ⅲ相国際共同試験:PULSAR試験]電子添文改訂時評価資料(96週)
3)バイエル薬品社内資料[薬物動態:母集団薬物動態解析]アイリーア8mg承認時評価資料
4)バイエル薬品社内資料[日本人を含む第Ⅲ相国際共同試験:PULSAR試験 継続期間]
